Got through to enriched biological processes for the 2021 data set. I also have some short-term goals of what I hope to have done before leaving for our sea star team writing retreat on Quadra on Saturday!
BLAST -ed Genome
I ran BLAST a while ago, but the output genes are transcripts (have the .t# suffix).
Code: paper-pycno-sswd-2021-2022/code/16-blast-genome-annotation.Rmd
BLAST output: paper-pycno-sswd-2021-2022/analyses/16-blast-annotation/blast_out_sep.tab
head blast_out_sep.tab
g3452.t1 sp Q9Y6A2 CP46A_HUMAN 36.842 380 201 6 73 1119 16 387 6.38e-73 238
g3453.t1 sp Q9Y6A2 CP46A_HUMAN 39.574 470 267 7 79 1470 20 478 2.82e-111 342
g3454.t1 sp Q9H4F8 SMOC1_HUMAN 41.026 156 80 3 43 504 21 166 3.34e-31 129
g3455.t1 sp Q9D844 DNJC4_MOUSE 35.028 177 100 3 100 618 37 202 1.38e-22 95.9
g3456.t1 sp P80018 GLBC_MOLAR 27.559 127 91 1 115 495 17 142 1.81e-08 54.7
g3457.t1 sp Q9NRC6 SPTN5_HUMAN 31.110 1649 1112 11 1 4926 1850 3481 0.0 758
g3458.t1 sp P16546 SPTN1_MOUSE 36.947 655 411 2 67 2028 300 953 1.10e-123 414
g3459.t1 sp Q00963 SPTCB_DROME 45.908 782 414 4 40 2361 36 816 0.0 691
g3460.t1 sp Q9WVK8 CP46A_MOUSE 38.248 468 266 9 103 1470 22 478 2.16e-106 329
g3461.t1 sp Q9Y6A2 CP46A_HUMAN 37.681 414 218 9 79 1254 22 417 2.44e-86 275
I asked in a GitHub issue what to do about this because I want to join the BLAST output with the DEG list for 2021 (listed below), but the files won’t have anything to join over, since the DEG list is genes (no .t# suffix).
So I’ll follow Sam’s advice:
Use existing BLAST output, but strip the .t1 from the first column and then sort for uniques just on the first column.
sed 's/\.t1//' blast_out_sep.tab | sort --unique -k1,1
This is probably fine, since it’s unlikely that multiple transcripts for a given gene would get annotated as a different protein.
Then, join with DEG list.
Fixed in the same R code file: paper-pycno-sswd-2021-2022/code/16-blast-genome-annotation.Rmd.
Here’s the new BLAST output:
head blast_out_sep_genes.tab
g10 sp Q94A76 GORK_ARATH 42.667 75 40 1 31 255 537 608 8.11e-07 49.7
g100 sp Q90WJ8 AJL2_ANGJA 29.861 144 92 4 70 474 20 163 3.68e-11 61.6
g1000 sp Q6AY85 ALG14_RAT 53.846 182 81 1 130 666 35 216 1.86e-73 224
g10000 sp Q8C163 EXOG_MOUSE 38.281 256 150 5 34 792 16 266 1.39e-52 178
g10002 sp Q9D1J3 SARNP_MOUSE 37.113 97 54 1 286 576 101 190 1.33e-12 67.8
g10003 sp Q4PJW3 CP51A_BOVIN 64.189 444 138 4 166 1443 60 500 0.0 604
g10004 sp P48449 LSS_HUMAN 63.800 721 260 1 13 2175 8 727 0.0 984
g10005 sp Q99996 AKAP9_HUMAN 36.364 418 191 10 1243 2463 3543 3896 6.97e-54 207
g10006 sp Q70FJ1 AKAP9_MOUSE 29.651 688 378 19 8368 10404 1678 2268 1.69e-36 157
g10007 sp Q6PB06 HYKK_XENLA 32.181 376 221 12 43 1140 12 363 4.38e-45 162
YAY!!!
Get Gene Ontology for BLAST Output
Following Steven’s class FISH 546 tutorial: here
I used the uniprot.org ID Mapping feature to get the Gene Ontology annotations.
I opened the BLAST output above in excel locally, copied all the uniprot accession IDs, and pasted them in the ID Mapping tool:
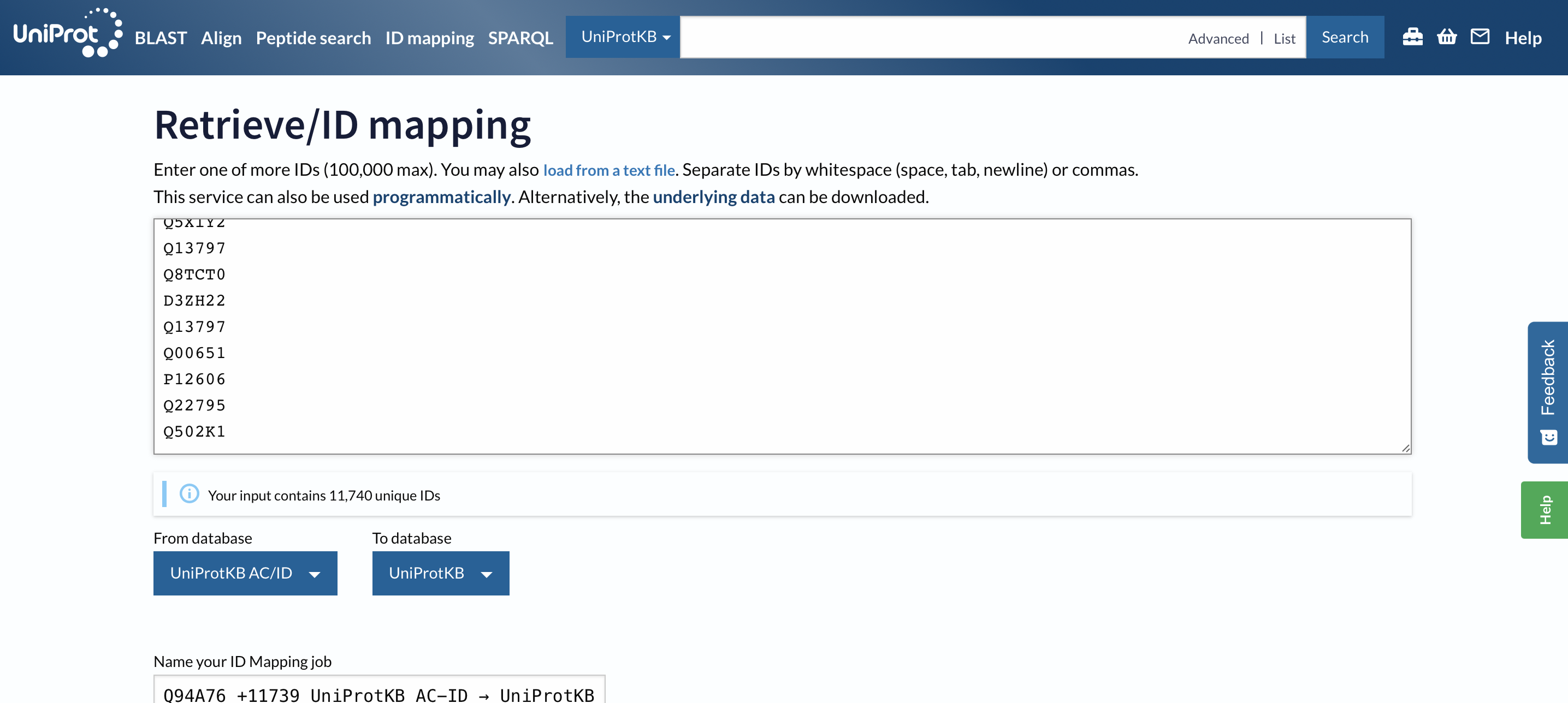
Select the start map button:
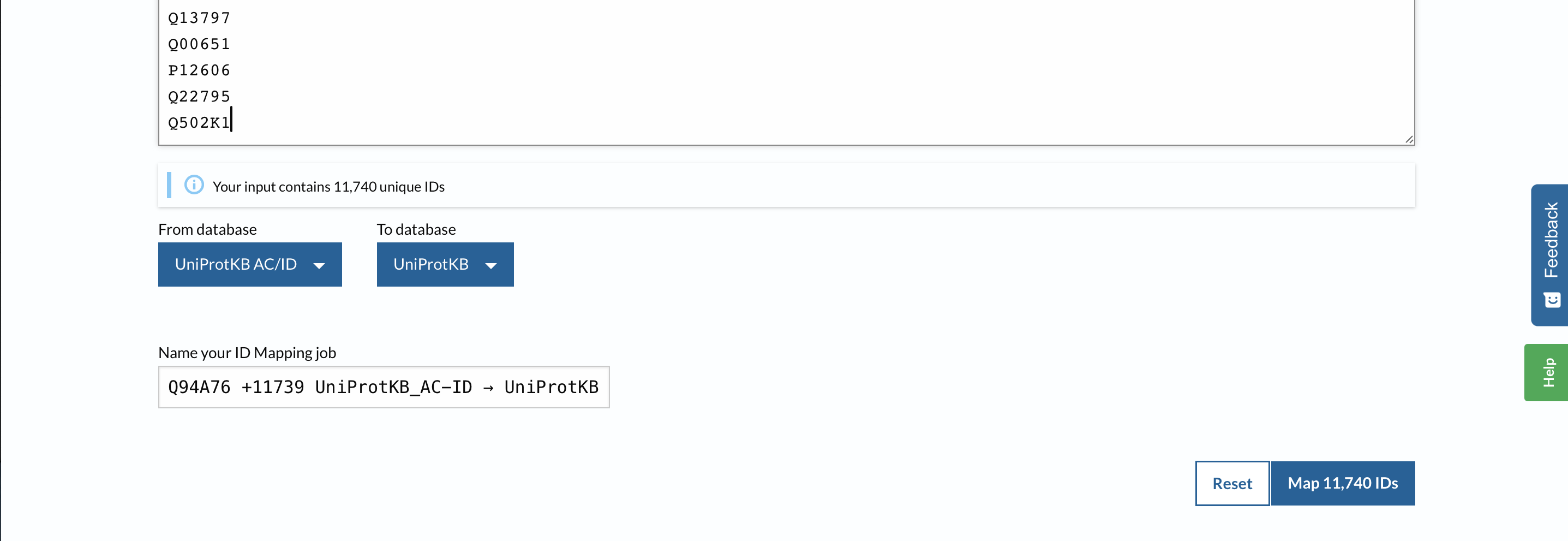
Mapping is complete, click on the word “complete”:
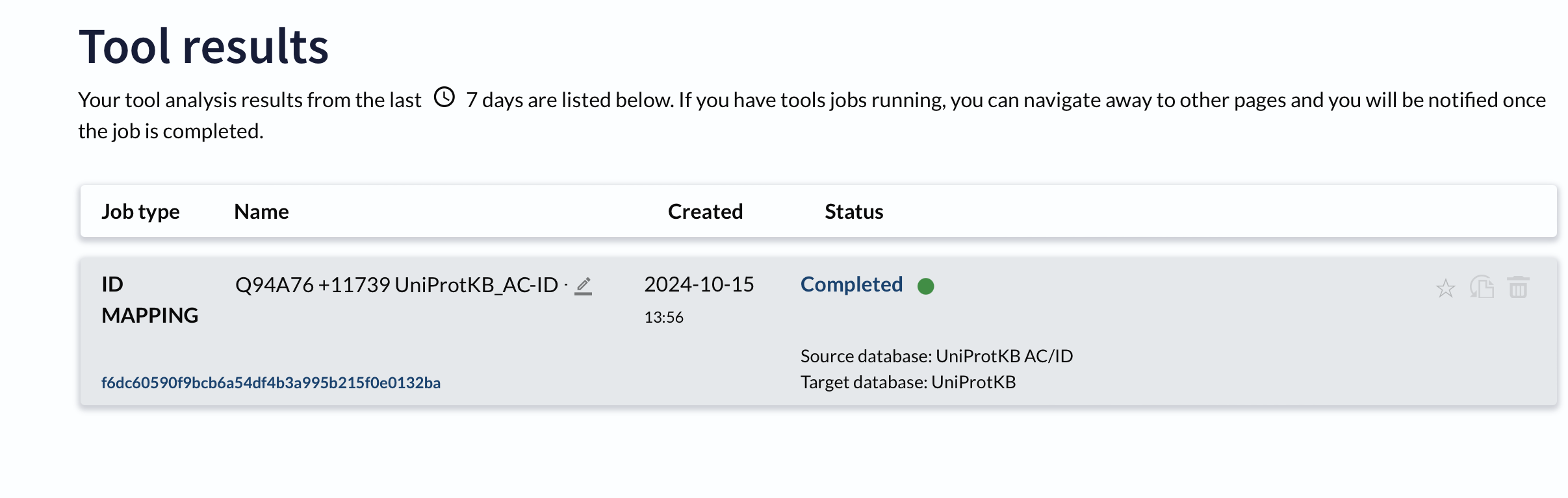
Select Customize Column option:
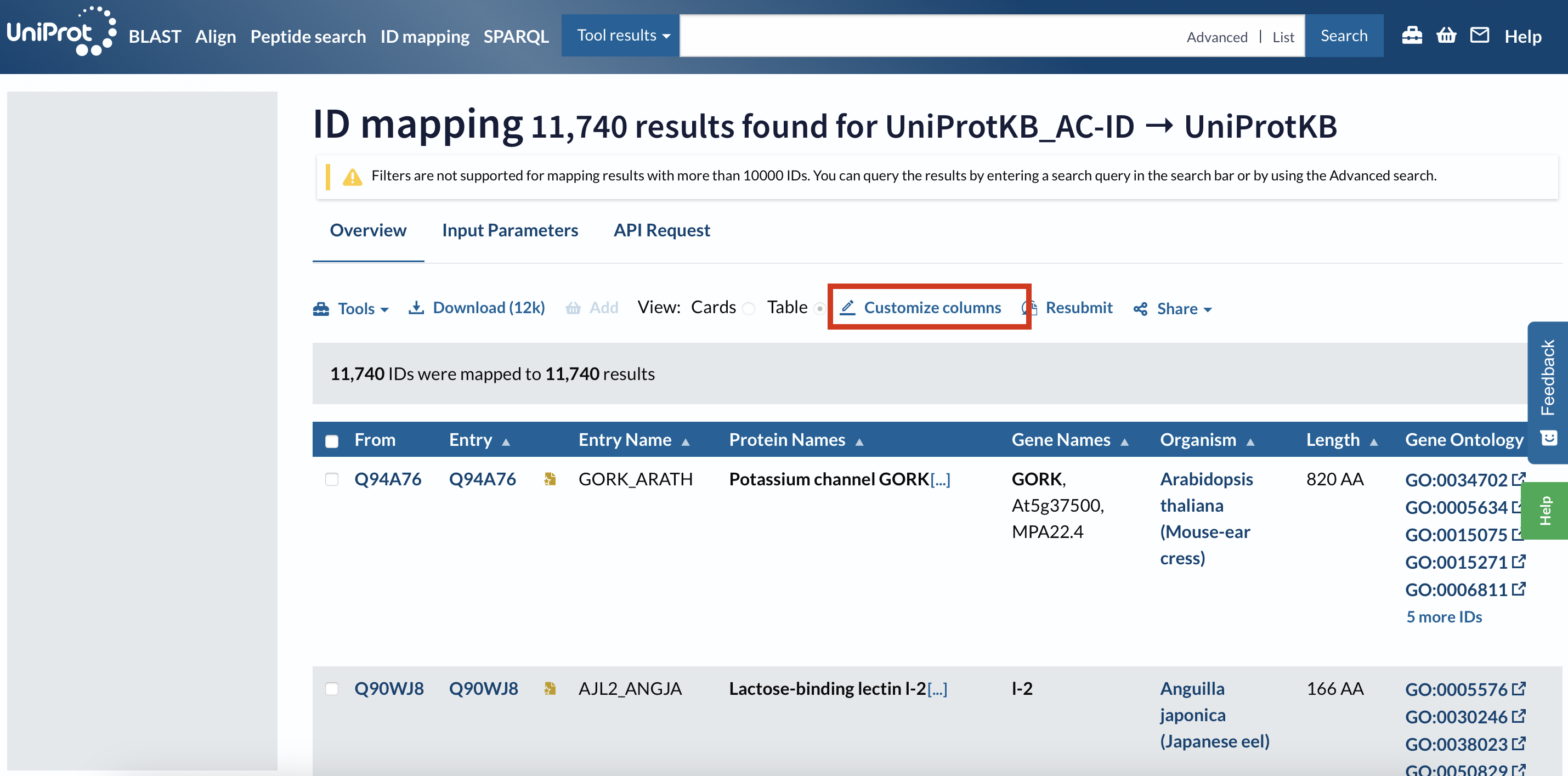
Select GO IDs:
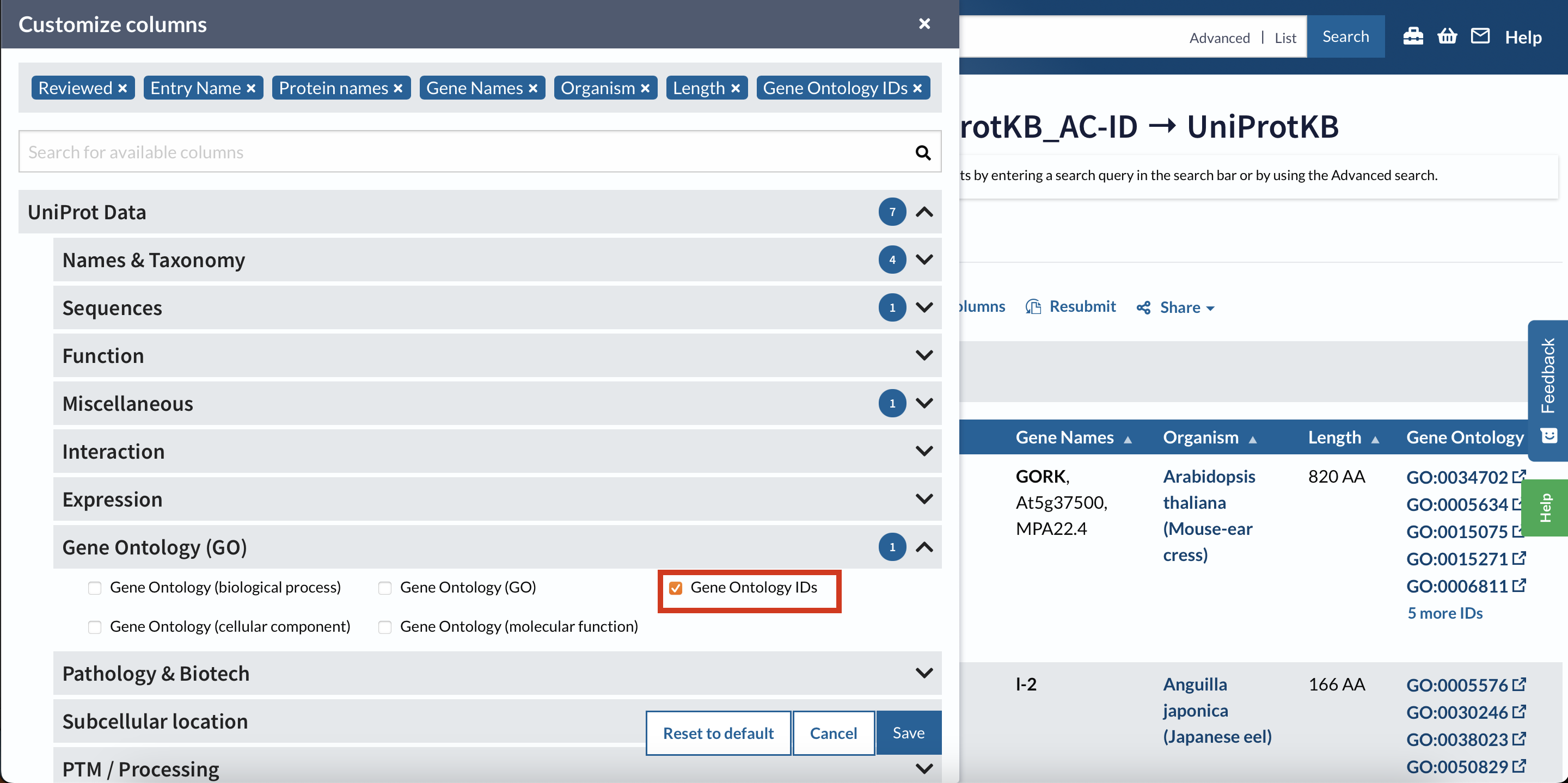
Then download the file as a .tsv.
Then copy and paste from downloads into paper-pycno-sswd-2021-2022/analyses/23-annotating-deg-lists. And push to GitHub.
2021 DEG list Annotation –> Enrichment
Summer 2021 DEG List Annotation
Ran DESeq2 to compare the 8 control star RNAseq libraries to the 8 exposed star RNAseq libraries. Got a list of DEGs (6,938), and annotated them by join-ing with the BLAST output from above.
Code using DESeq2: paper-pycno-sswd-2021-2022/code/21-deseq2-2021.Rmd
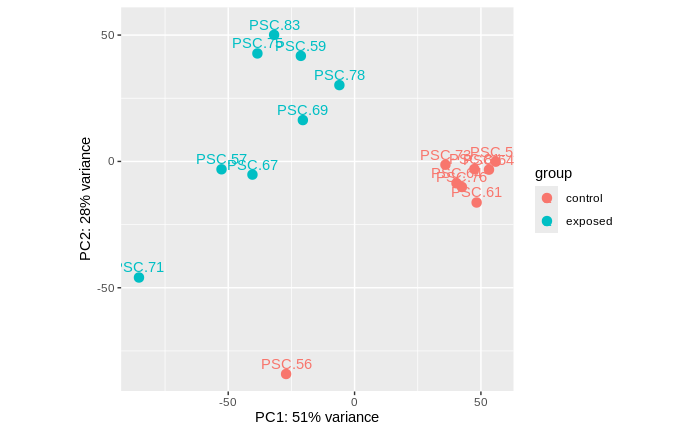
PCA of the libraries compared:
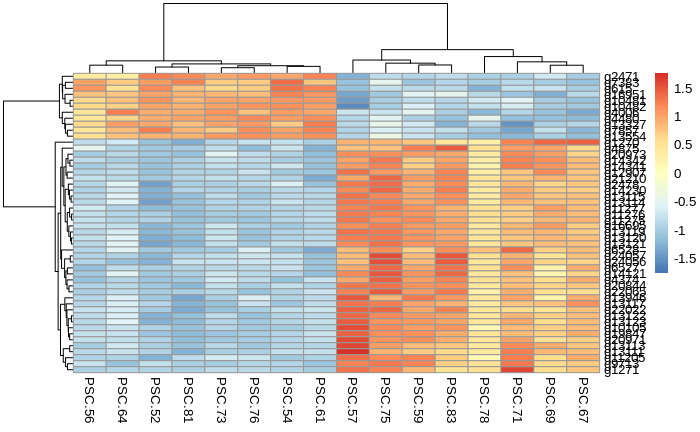
Heatmap of the top 50 DEGs from the comparison:
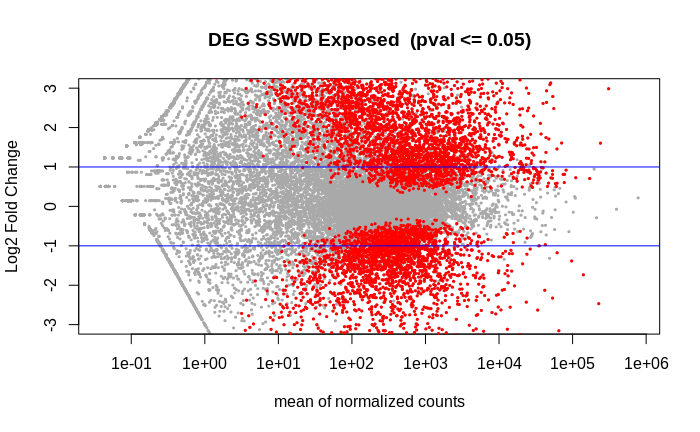
Volcano of the 6938 DEGs:
DEG list: paper-pycno-sswd-2021-2022/analyses/21-deseq2-2021/DEGlist_2021_exposedVcontrol.tab
head DEGlist_2021_exposedVcontrol.tab
baseMean log2FoldChange lfcSE stat pvalue padj
g21712 15506.8006860189 -0.686001394104519 0.279769093821729 -2.45202707966608 0.0142053971059541 0.04104326062556
g21713 931.361342578574 0.765760120416294 0.279988740054853 2.73496755714631 0.00623864245462093 0.0207814457271467
g21711 12883.0935253533 -0.709970512692711 0.228673239474894 -3.10473807220743 0.00190447593029036 0.00763473087082639
g21769 95.2608451733162 3.6958184007692 1.00771829984027 3.66751144774787 0.000244922588350467 0.00136039062124746
g15181 7362.04249036577 2.13117880973538 0.606155663157497 3.5158935885115 0.000438276606665117 0.00222160900619904
g15182 848.103634775757 1.44107523921733 0.603474139884921 2.38796519017722 0.0169419463881676 0.047565397521059
g15183 916.675431757199 -0.700558231713056 0.24491415606803 -2.86042359886482 0.00423075479942892 0.0149434418878582
g7651 625.045813674949 1.17515444248357 0.338850532449784 3.46806137203791 0.000524227551336494 0.00257724022366272
g7656 369.553524158447 1.34279754324671 0.359840419468955 3.73164733752917 0.000190231694582979 0.00109975735292238
2,934 DEGs are more expressed in the controls (~42%)
4,004 DEGs are less expressed in the controls (~58%)
I got these numbers in excel… I averaged the counts for the genes in the controls (n=8 libraries), and averaged the counts for the genes for the exposed (n=8), and if the value was higher in the controls, a column was populated with “yes”, and if the value was less in controls compared to the exposed, the column was popuulated with “no”.
Annotate DEG list
Code to annotate DEG list: paper-pycno-sswd-2021-2022/code/23-annotating-deg-lists.Rmd
Annotated DEG list: paper-pycno-sswd-2021-2022/analyses/23-annotating-deg-lists/DEGlist_2021_exposedVcontrol_annotated.tab
head -3 DEGlist_2021_exposedVcontrol_annotated.tab
gene_id baseMean log2FoldChange lfcSE stat pvalue padj PSC.56 PSC.52 PSC.54 PSC.61 PSC.64 PSC.73 PSC.76 PSC.81 PSC.59 PSC.57 PSC.69 PSC.67 PSC.71 PSC.75 PSC.78 PSC.83 Vuniprot_accession gene_name V5 V6 V7 V8 V9 V10 V11 V12 V13 V14 Entry Reviewed Entry.Name Protein.names Gene.Names Organism Length Gene.Ontology.IDs
1 g21712 15506.8006860189 -0.686001394104519 0.279769093821729 -2.45202707966608 0.0142053971059541 0.04104326062556 16897 26645 32348 20860 32914 28245 21628 26422 10051 7784 7655 8444 8324 2531 22624 6679 sp P54985 PPIA_BLAGE 73.171 164 44 0 1 492 1 164 2.17e-88 258 P54985 reviewed PPIA_BLAGE Peptidyl-prolyl cis-trans isomerase (PPIase) (EC 5.2.1.8) (Cyclophilin) (Cyclosporin A-binding protein) (Rotamase) CYPA Blattella germanica (German cockroach) (Blatta germanica) 164 GO:0003755; GO:0005737; GO:0006457; GO:0043231
2 g21713 931.361342578574 0.765760120416294 0.279988740054853 2.73496755714631 0.00623864245462093 0.0207814457271467 1025 718 596 910 905 905 1269 1044 1602 1167 479 2113 1073 128 584 1016 sp Q8TDM6 DLG5_HUMAN 34.563 732 353 15 310 2409 158 795 1.74e-107 387 Q8TDM6 reviewed DLG5_HUMAN Disks large homolog 5 (Discs large protein P-dlg) (Placenta and prostate DLG) DLG5 KIAA0583 PDLG Homo sapiens (Human) 1919 GO:0001837; GO:0005737; GO:0005886; GO:0005912; GO:0007165; GO:0008013; GO:0008092; GO:0008285; GO:0014069; GO:0030011; GO:0030054; GO:0030159; GO:0030336; GO:0030859; GO:0030901; GO:0035331; GO:0035332; GO:0035556; GO:0036064; GO:0042130; GO:0042981; GO:0045176; GO:0045186; GO:0045197; GO:0045880; GO:0051965; GO:0060441; GO:0060563; GO:0060999; GO:0065003; GO:0071896; GO:0072205; GO:0098609; GO:0098978; GO:0099173
Summer 2021 Enrichment
Using DAVID and REVIGO.
R code to get lists for DAVID: paper-pycno-sswd-2021-2022/code/24-2021-2022-enrichment.Rmd
DAVID
Background (genome BLAST uniprot accession IDs):
paper-pycno-sswd-2021-2022/analyses/24-2021-2022-enrichment/blast_uniprotAccession_forDAVID.txt
Gene Ontology Biological Processes results:
DAVID output:
paper-pycno-sswd-2021-2022/analyses/24-2021-2022-enrichment/DAVID_2021_DEGlist.txt
I’m not sure what the best metric is for looking at enrichment from DAVID.
Kegg Pathway list:
paper-pycno-sswd-2021-2022/analyses/24-2021-2022-enrichment/DAVID_2021_KEGG_pathway.txt
I found this youtube video on how to use DAVID, that basically confirms that I’ll be using the GOTERM_BP_DIRECT list (which is what’s linked above), and the KEGG pathways. I’m not totally sure which metric is best for identifying the most enriched processes - whether that be Bonferroni, FDR, p-value, etc. So I’ll look into that this week.

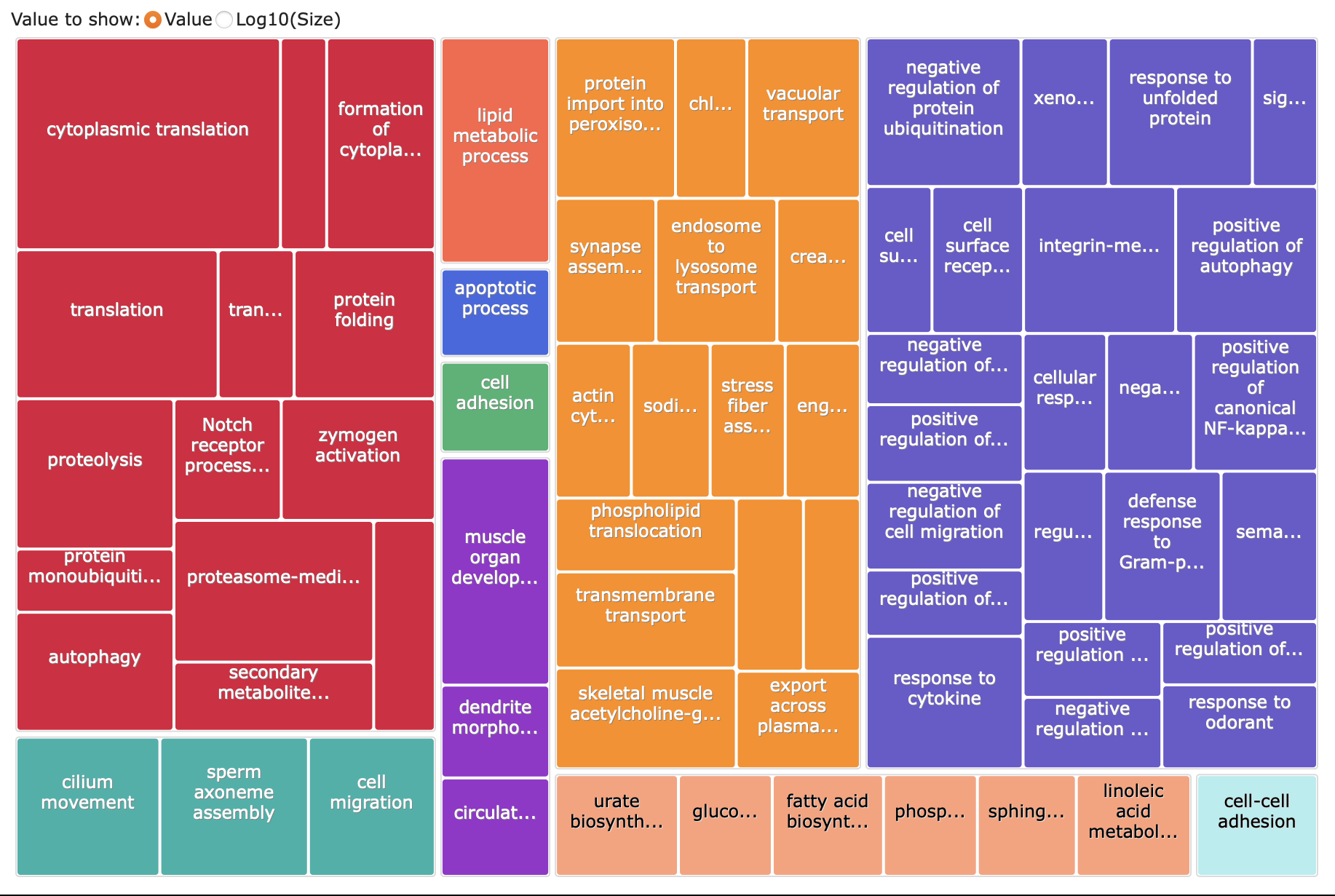
REVIGO
Here’s what I put into REVIGO: paper-pycno-sswd-2021-2022/analyses/24-2021-2022-enrichment/2021_GO_pvalue_for_REVIGO.txt
It’s the GO terms from the DAVID output for the terms that had a p-value < 0.05.
Here’s the treemap of that analysis:
Finding Cool Genes
What I can do from here, is look at the GO IDs from the enriched terms in paper-pycno-sswd-2021-2022/analyses/24-2021-2022-enrichment/2021_GO_pvalue_for_REVIGO.txt, and then find that GO ID in the 2021 annotated DEG list, and find the genes associated with that term. There are going to be multiple/many genes associated with each GO ID, so I’ll pick the ones that are related to stress/imumune response.
I can then look at the counts of that gene across the libraries and see if it’s more expressed in controls vs exposed. I can also create a visual of those counts across samples with a heatmap.
Short-term Goals
Get analyses for 2021 and 2022 solidly done by Friday end of day.
This means:
2021
- DEG list annotation (DONE)
- Enrichment DAVID output (DONE)
- REvigo images
- Genes of interest highlighted
- Papers related to genes of interest compiled
2022
- DEG lists annotated
- Lists: - control vs exposed - control vs exposed with age into account - age contrast (just DEGs influenced by age that are related to control vs exposed)
- Enrichment DAVID outputs
- Revigo images
- Genes of interest highlighted
- Papers related to genes of interest compiled