Today I extracted RNA from 10 more pelleted hemolymph samples using Tri-Reagent (without lyophilizing). I ran 5 ul on the RNA HS Qubit, and 7/10 had quantifiable RNA. I also started working with the .fastq sequence data from our first sequenced library. I ran the four files through FASTQC and am learning how to determine if the output is good.
RNA Isolation
Today I isolated the following samples using the protocol outlined here.
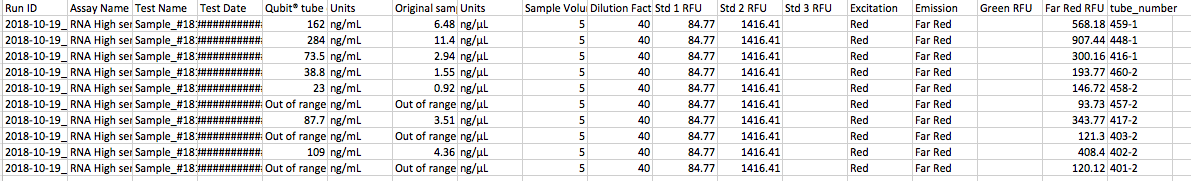
70% success is not bad!
On Monday, Steven, Sam, and I are going to discuss how to move forward. The Bioanalyzer results from the first time I extracted RNA using Tri-reagent and not lyophilizing did not look great, so we’ll have to see how to proceed. The lyophilizer is still broken.
Starting to work with .fastq data
We received notice on Oct. 15th that our data from our first library!
Sam did some work with it, but I’ll be replicating some of it before I continue on. Currently, I’m working on running the four .fastq files through FASTQC.
Here are url’s to my output files:
http://htmlpreview.github.io/?https://github.com/fish546-2018/grace-Cbairdi-transcriptome/blob/master/analyses/304428_S1_L001_R1_001_fastqc.html http://htmlpreview.github.io/?https://github.com/fish546-2018/grace-Cbairdi-transcriptome/blob/master/analyses/304428_S1_L001_R2_001_fastqc.html http://htmlpreview.github.io/?https://github.com/fish546-2018/grace-Cbairdi-transcriptome/blob/master/analyses/304428_S1_L002_R1_001_fastqc.html http://htmlpreview.github.io/?https://github.com/fish546-2018/grace-Cbairdi-transcriptome/blob/master/analyses/304428_S1_L002_R2_001_fastqc.html
I will be using this data for my FISH546 project.
Some things I learned today/next steps notes:
- There are so many files in /nightingales/C_bairdi/ even though it is from 1 pooled sample because the sample was run in two lanes, and there was a reverse and a forward run.
- Next steps after FASTQC:
- Combine files into one (concatenate)
- Trim (if necessary)
- Assemble
This weekend and Monday I’ll play around with Trinity. Tuesday in FISH546 class we’ll go into the details of the FASTQC output to see what they mean.