Today I discussed with Sam about how to use R for making a better master file. We identified some short term and long term goals for me to work on (and get help with via GitHub Issues). I also ran the Bioanalyzer on Test3 (from when lyophilizer was used and using Tri-reagent ), and four samples that I extracted last Friday using (wihtout lyophilizer, with Tri-reagent). The Bioanalyzer didn’t run great because the ladder and the markers didn’t show up… will have to re-run.
R code goals
Long-term:
- Create a code for making a hemolymph data sheet.
Currently, my hemo master data (20180522-all-crabs-hemo.csv) is copy and pasted from an excel file that Pam sent me. It’s copied from the tab labeled “all crabs”. The excel sheet has empty cells, so in order to make the .csv, I manually filled the cells.
Short-term:
- Create code for reading in new Qubit files, creating a vector of the tube_numbers
- figure out how to make the vector to join the Qubit data file (make it a column called “tube_number”)
- add the “extraction_method” to the new Qubit file
- add yields in ng
- join the new Qubit file to the hemosample_qubit_table.csv based on “tube_number”
Having a workflow for this will make my life SO MUCH easier, as I have a lot more RNA extractions to do, and I have to keep track of which samples have been extracted, what the yields were, and extraction method.
Bioanalyzer results
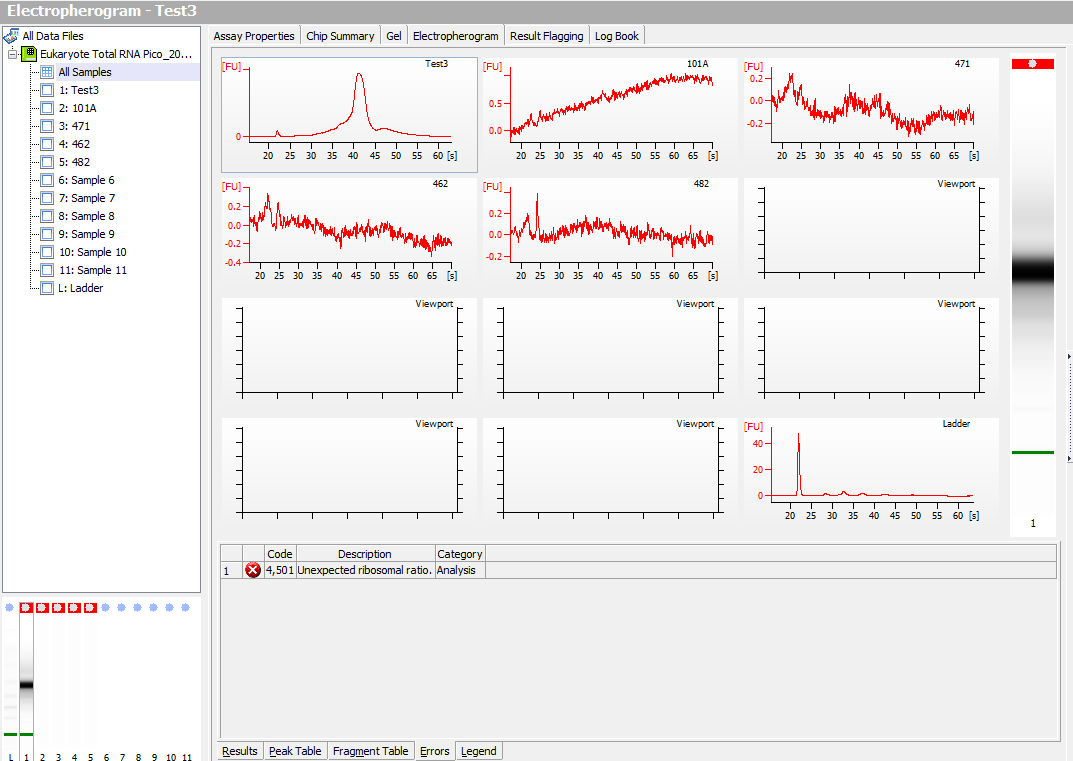
I ran five samples in the Bioanalyzer.
Test 3, one of the lyophilized samples from what Sam extracted (the other two didn’t have detectable RNA in the Qubit, so Sam only gave me the sample with detectable RNA) using Tri-reagent protocol.
The other four samples are from what I extracted (not lyophilized) using Tri-reagent protocol on Friday.
Electropherogram:
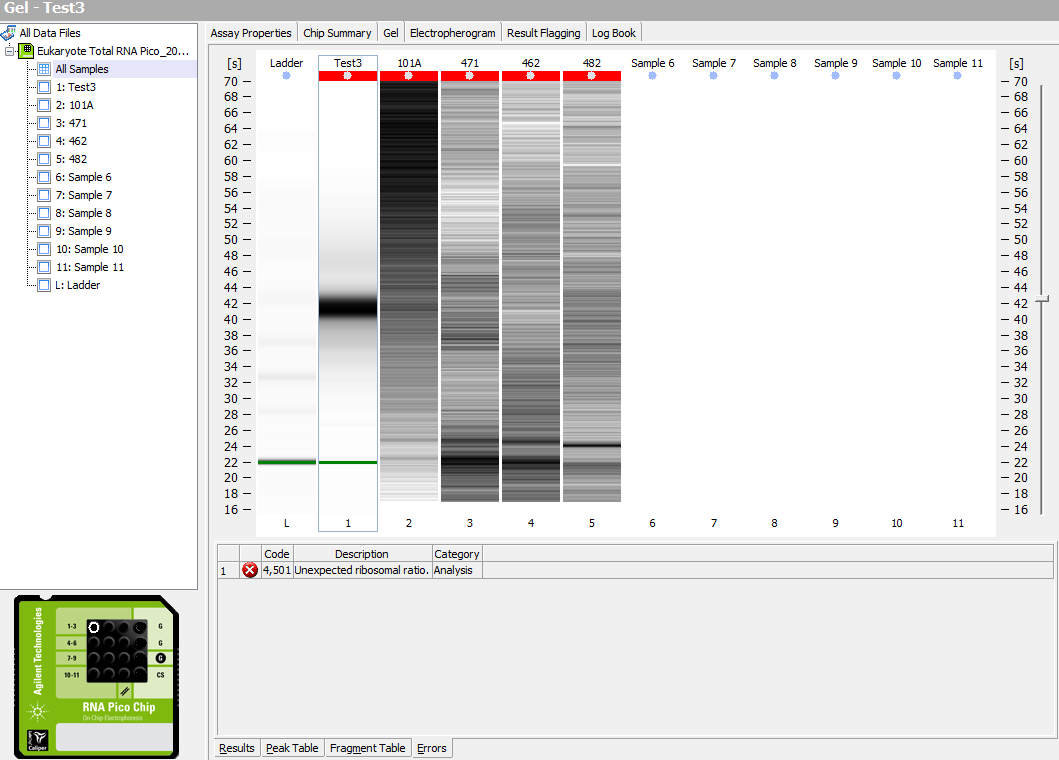
Gel:
Thoughts on the results:
Clearly, the samples that I extracted on Friday were no good, even though they had detectable RNA.
The ladder didn’t show up, so that’s not good… I’ll have to re-run.
GitHub Issue #393